Roles of Claspin in regulation of DNA replication, replication stress responses and oncogenesis in human cells
Review Article
Open Access
Genome Instability & Disease (2021)
Abstract
Human cells need to cope with the stalling of DNA replication to complete replication of the entire genome to minimize genome instability. They respond to “replication stress” by activating the conserved ATR-Claspin-Chk1 replication checkpoint pathway. The stalled replication fork is detected and stabilized by the checkpoint proteins to prevent disintegration of the replication fork, to remove the lesion or problems that are causing fork block, and to facilitate the continuation of fork progression. Claspin, a factor conserved from yeasts to human, plays a crucial role as a mediator that transmits the replication fork arrest signal from the sensor kinase, ataxia telangiectasia and Rad3-related (ATR), to the effector kinase, Checkpoint kinase 1 (Chk1). Claspin interacts with multiple kinases and replication factors and facilitates efficient replication fork progression and initiation during the normal course of DNA replication as well. It interacts with Cdc7 kinase through the acidic patch segment near the C-terminus and this interaction is critical for efficient phosphorylation of Mcm in non-cancer cells and also for checkpoint activation. Phosphorylation of Claspin by Cdc7, recruited to the acidic patch, regulates the conformation of Claspin through affecting the intramolecular interaction between the N- and C-terminal segments of Claspin. Abundance of Claspin is regulated at both mRNA and protein levels (post-transcriptional regulation and protein stability) and affects the extent of replication checkpoint. In this article, we will discuss how the ATR-Claspin-Chk1 regulates normal and stressed DNA replication and provide insight into the therapeutic potential of targeting replication checkpoint for efficient cancer cell death.
Introduction
Cancer incidence is a prevalent cause for human death in developed countries with aging society. How to effectively inhibit cancer cells from progression, metastasis and even relapse is one of the key medical issues that are being studied in different disciplines from various points of views (Klein, 2020). Cancers can arise from accumulation of genetic alterations that may be generated during chromosome replication and inheritance (Andor et al., 2017). Accordingly, investigation of molecular mechanisms of maintenance of genome integrity has been a mainstay of cancer research.
The major cause of genome instability is the presence of replication stress during DNA replication which stalls replication fork progression and reduces the replication fork rate (Bartkova et al., 2006; Di Micco et al., 2006; Gorgoulis et al., 2005). The sources of replication stress include lesions on DNA (DNA strand breaks, double-strand cross-linking, DNA assaults, and chemical modification of bases, etc.), unusual DNA structures on the template DNA, reduced supply of nucleotide precursors, and collision of replication and transcription. The replication stress can be induced by untimely induction of DNA replication or by dysregulated origin firing (Gaillard et al., 2015).
To preserve the genome integrity during replication stress, the ATR-Claspin-Chk1 replication checkpoint pathways is activated (Gaillard et al., 2015). Here, we will mainly focus on the cellular roles of the ATR-Claspin-Chk1 pathway in control of DNA replication, replication stress responses and oncogenesis. Readers are also referred to other recent reviews on Claspin (Azenha et al., 2019; Smits et al., 2019).
Functional roles of Claspin during normal DNA replication, replication fork progression and initiation
Although Claspin and its yeast ortholog, Mrc1, were discovered as a regulator of replication checkpoint, they have been shown to regulate origin firing and fork progression during the normal course of DNA replication both in yeast and human cells (Hayano et al., 2011; Petermann et al., 2008; Yeeles et al., 2017). Recently, we reported that Claspin is required also for DNA replication initiation in non-cancer cells (Masai et al., 2017; Yang et al, 2016). Claspin recruits Cdc7 kinase, essential for initiation, to the conserved acidic patch (AP) region (residues 986–1100) near the C-terminus of Claspin. The replacement of the acidic residues in AP with alanine impairs the Cdc7 binding and abolishes the ability of Claspin to recruit Cdc7 kinase, and reduces the phosphorylation of Mcm (Fig. 1A). Accordingly, the rate of DNA synthesis is compromised in the Claspin AP-mutant MEF cells. Cdc7 can also phosphorylate multiple sites on Claspin (Kim et al., 2008; Rainey et al., 2013; Yang et al., 2019). Indeed, DE/A mutant of Claspin is not phosphorylated by Cdc7 kinase in vitro. The AP mutation abrogates the intramolecular looping between the N-terminal (N-ter) and C-terminal (C-ter) regions within Claspin. This intramolecular interaction suppresses the DNA- and PCNA-binding activities of Claspin, and is disrupted by phosphorylation of the N-ter by Cdc7, strongly suggesting that the recruitment of Cdc7 by Claspin plays important roles in initiation not only by facilitating the phosphorylation of Mcm proteins, but also by activating DNA and PCNA binding of Claspin (Masai et al., 2017; Matsumoto et al., 2017; Yang et al, 2016).
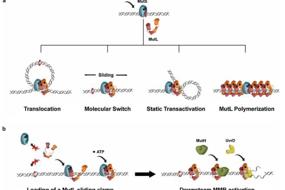
Fig. 1
The roles of Claspin in DNA replication initiation and fork progression. A The AP (Acidic Patch) and basic patch segments of Claspin undergo intramolecular interaction. Cdc7 kinase is recruited to AP of Claspin on the chromatin (indicated by a red arrow; ①), facilitating the phosphorylation of Mcm and Claspin itself. (indicated by green arrows) This phosphorylation disrupts the intramolecular interactions within Claspin (②) and facilitates its binding to PCNA (indicated by a red arrow) and DNA (③). All of them contribute to efficient DNA replication initiation. B Through direct interactions with Tipin, Timeless, and CMG complex, Claspin stabilizes the replication fork, regulates fork speed, and ensures efficient fork progression
Full size imageTo ensure appropriate replication fork progression, Ctf4 (And-1), Mrc1 (Claspin), and Csm3/Tof1 (Tipin/Timeless) together with CMG complexes, constitute replisome progression complexes in yeast (Baretić et al., 2020; Gambus et al., 2006; Fig. 1B). Claspin has been shown to be an integral component of the replisome progression complex, and loss of Claspin leads to reduced replication fork rate in cells. The direct role of Mrc1 in efficient DNA replication fork progression in yeast has been further demonstrated using the in vitro reconstituted DNA replication system (Yeeles et al., 2017). In the reconstituted DNA replication system with a set of origin firing proteins (e.g., Cdc45, DNA polymerase ε, Mcm10, Sld3/7, Sld2, Dpb11, S-CDK, GINS), replication elongation factors (e.g., TopoII, DNA polymerase α, RPA, Ctf4) and fork-associating factors (Mrc1, CMG, FACT, TopoI, Csm3, and Tof1), omission of Mrc1 dramatically decreases DNA synthesis rate, nearly identical to the replication profile exhibited by minimal replisome which is composed of all origin firing proteins along with TopoII and Ctf4. Replication fork rate is much reduced in the absence of Csm3 and Tof1, suggesting Mrc1 in conjunction with Csm3 and Tof1 contributes to replication fork progression at the maximum rate (Yeeles et al., 2017).
It was previously reported that the interaction between Mrc1 and Hsk1 (fission yeast homologue of Cdc7 kinase) regulates origin firing in yeast in a checkpoint-independent fashion (Matsumoto et al., 2017). This interaction relies on the Hsk1 bypass segment (HBS) in Mrc1 to which Hsk1 binds. Similar to Claspin that is phosphorylated by Cdc7, Mrc1 is robustly phosphorylated by Hsk1 kinase through the HBS and this phosphorylation leads to the disruption of intramolecular looping formed between HBS and N-terminal target of HBS (NTHBS) within Mrc1 (Matsumoto et al., 2017). Thus, regulation of Claspin/Mrc1 through an intramolecular interaction is evolutionally conserved.
Furthermore, Claspin (Mrc1 in yeast) in conjunction with TIMELESS (Tof1 in yeast) and TIPIN (Csm3 in yeast) is physically tethered to mini-chromosome maintenance (MCM) DNA helicase subunits and DNA polymerases. This association is important for both efficient fork progression and for fork stabilization under normal and replication stress conditions (Leman & Noguchi, 2012; Leman et al., 2010; Petermann et al., 2008; Yoshizawa-Sugata & Masai, 2007). Recent studies show that Mrc1 forms a complex called MTC or fork protection complex with Tof1 and Csm3 (Lewis et al., 2017; Noguchi et al., 2003). The association of the MTC complex with the fork significantly increases the fork speed and replication progression, shown by DNA stretching assay at the single-molecule level. The transient interaction between MTC and replisome is due to the weak affinity of MTC to replisomes, making the movement of the replisome a highly dynamic process. It has also been reported that Mrc1 in cooperation with Tof1 and Csm3 protects specifically CAG repeats from DNA contractions and breakages. This is indicated by the finding that Mrc1 deletion leads to higher vulnerability of CAG repeats (Gellon et al., 2019). The protective functions of Mrc1 in CAG repeat stability might be due to the fact that the MTC complex can be coupled to the helicases, such as Srs2 and Sgs1, in yeast. Similar roles of Claspin-Timeless-Tipin in stabilization of trinucleotide repeats are observed in human cells (Liu et al., 2012a, 2012b).
Although Mrc1 and Claspin are orthologs and possess functional and mechanical similarities including intramolecular regulation between N- and C-terminal segments, as stated above, there may be some differences in terms of their functional significances (Matsumoto et al., 2017). The N–C interaction in the yeast Mrc1 appears to contribute to the negative regulation of initiation, since its disruption causes precocious initiation specifically at early-firing origins. In mammalian cells, this interaction contributes positively to the initiation in non-cancer cells. However, the effects of the intramolecular interaction differ between cell types in mammalian cells (Hsiao et al. unpublished data), and thus roles of Claspin need to be evaluated in various cell types. Claspin could also negatively regulate the initiation in mammalian cells (see below).
The conserved ATR-Claspin-Chk1 pathway regulates replication checkpoint
The stalled replication forks are quickly detected by the conserved ATR-Claspin and various downstream events are induced, including the suppression of origin firing, slowed replication fork progression and inhibition of mitosis (Tercero et al., 2003). However, stalled replication forks could generate DNA breaks (nicks) if they are not properly attended by the ATR-Claspin-Chk1 checkpoint pathway. In yeast, the DNA replication checkpoint kinase, Cds1, suppresses the endonuclease activity of Mus81/Eme1 during perturbed DNA replication to maintain genome stability (Froget et al., 2008). The Mus81/Eme1 heterodimeric complex can cleave the branched DNAs in a structure-specific manner. When the Cds1 functions improperly or is absent, DNA is cleaved by Mus81/Eme1 and stalled replication forks are at a higher risk of DNA breaks and mutations or more vigorous genomic rearrangements (Froget et al., 2008).
DNA damages are caused by persistent endogenous and exogenous genotoxic insults and activate DNA damage responses (DDRs) (Gaillard et al., 2015; Técher et al., 2017). Replication checkpoint/DNA damage checkpoints further activate subsequent checkpoint signaling pathways for DNA repair and cell cycle arrest. The ATR/Claspin/Chk1 in human or Mec1(Rad3)/Mrc1/Rad53(Cds1) in yeasts, well-conserved in eukaryotes, plays a crucial role in replication checkpoint control in response to replication stress (Bacal et al., 2018; Berens & Toczyski, 2012; Smits et al., 2019; Yang et al., 2019). However, how this axis is precisely controlled is not completely understood.
When replication fork is stalled, ssDNA regions are generated and are coated with replication protein A (RPA), which is then sensed by ATR-interacting protein (ATRIP) that forms a complex with ATR, triggering ATR auto-phosphorylation at Thr1989 (Liu et al., 2011). Then, the ATR–ATRIP complex stimulated by TopBP1 and ETAA1 phosphorylates Claspin, resulting in Chk1 phosphorylation at Ser317 and Ser345 which is mediated by ATR (Kim et al., 2008; Liu et al., 2011; Rao et al., 2018; Yang et al., 2019; Zou, 2017). Meanwhile, the stalled replication forks generated by replication stress are protected and stabilized by the complex containing Tim, TIPIN, Claspin and AND-1 (Kemp et al., 2010; Leman & Noguchi, 2012; Leman et al., 2010; Rageul et al., 2020; Yoshizawa-Sugata & Masai, 2007). As previously described, Tim and Tipin are involved in the maintenance of replication forks under both normal replication and replication stress conditions (Leman & Noguchi, 2012; Leman et al., 2010; Yoshizawa-Sugata & Masai, 2007). In the presence of replication stress, Tipin interacts with RPA bound to ssDNA and regulates Claspin-dependent Chk1 phosphorylation (Kemp et al., 2010) (Fig. 2). It has been further shown that SDE2, a PCNA-interacting protein regulating DNA replication fork progression, is involved in Chk1 activation (Rageul et al., 2020). Since the recruitment of Claspin to chromatin is disrupted upon SDE2 or Tim depletion, it has been concluded that both SDE2 and Tim are required for Claspin engagement in the replication fork complex upon replication stress (Rageul et al., 2020).
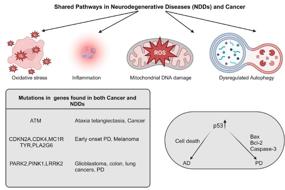
Fig. 2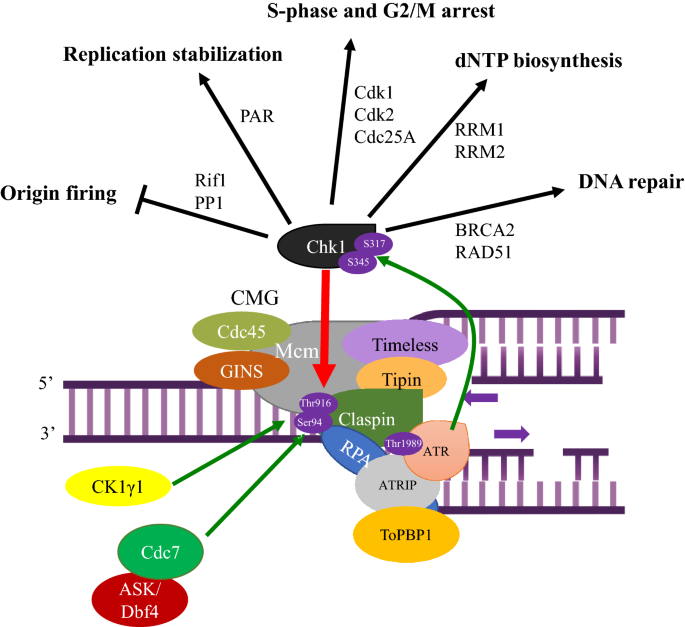
The signaling cascade of the ATR-Claspin-Chk1 replication checkpoint pathway. Upon fork stall caused by replication stress, ATR is activated by RPA/ATRIP (ATR-interacting protein) and TopBP1. Recruitment of ATR–ATRIP to RPA-coated ssDNA leads to auto-phosphorylation at Thr-1989 of ATR (Liu et al., 2011). Cdc7 (or CK1γ1) is recruited to Claspin (red arrow), phosphorylating T916 and S945 in CKBD of Claspin (indicated by green arrows), inducing the binding of Chk1, which is phosphorylated by ATR at S317 and S345 (indicated by a green arrow). Phosphorylated Chk1 now regulates origin firing, replication fork progression, cell cycle progression, repair of lesions and removal of the causes of replication stress. See text for more details
Full size imageTo sum up, Claspin is tightly associated with successful replication checkpoint activation in response to replication stress through a complex protein regulatory network. Although a number of proteins have been identified to regulate the ATR-Claspin-Chk1 axis, the detailed molecular mechanisms require further investigation.
Regulation of Claspin expression and stability in its functional control
Stabilization and modification of Claspin at both transcriptional and post-translational levels play a crucial role in replication checkpoint signaling in response to replication stress. The Claspin mRNA can be structurally stabilized by tristetraprolin (TTP), mRNA-interacting protein (Lee et al., 2020) (Fig. 3A). The association of 3’ untranslated region of Claspin and TTP is required for Claspin to exert its normal functions, including replication fork progression and appropriate replication checkpoint activation. This is indicated by the fact that TTP depletion significantly thwarts normal DNA replication fork progression and diminishes Claspin-dependent Chk1 phosphorylation after replication stress.
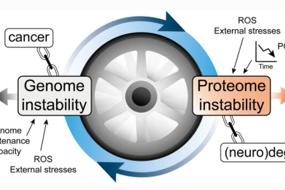
Fig. 3
Claspin stability during cell cycle is regulated at the mRNA and protein levels. A Effect of TTP-mediated regulation on the Claspin mRNA during S phase. In S phase, Claspin mRNA can be stabilized via binding of TTP to its 3’ UTR. B Effect of post-translational modifications on Claspin stability during cell cycle. Claspin can be targeted and regulated by ubiquitin-mediated proteasomal degradation during unperturbed cell cycle progression
Full size imageAt the post-translational level, Claspin can be targeted and ubiquitinated by a series of ubiquitinases for its protein turnover and regulation during cell cycle progression. However, this can be antagonized by ubiquitin-specific peptidases (USPs), including USP7, USP9X, USP11, USP20, USP28, and USP29 (Faustrup et al., 2009; Ito et al., 2018; Martin et al., 2015; McGarry et al., 2016; Wang et al., 2017; Yuan et al., 2014; Zhao et al., 2021) (Fig. 3B). Depletion of hepatocyte nuclear factor 1-β (HNF-1β) compromises Claspin-dependent replication checkpoint activation, including Chk1 phosphorylation, in response to replication stress (Ito et al., 2018). This is because HNF-1β is needed to stabilize Claspin by stimulating USP28 gene expression and enhancing USP28-mediated deubiquitination. These findings strongly suggest that manipulation of Claspin expression and stability at both mRNA and post-translational levels can be exploited for controlling replication checkpoint activity.
Claspin stability is modulated by MTA1, a chromatin remodeler that drives transcriptional regulation, as well (Li et al., 2010). MTA1 is stabilized by UV which suppresses MTA1 ubiquitination and thus prevents its degradation, and is required for the efficient expression of Chk1 and Claspin. MTA1 interacts with ATR in response to UV, and maintains the expression levels of Claspin and Chk1, and thus is required for efficient replication checkpoint activation.
Protein kinases control replication checkpoint in response to biological stresses
The key step for the replication checkpoint signaling in response to replication stress and fork stalling is the recruitment of multiple protein kinases to the stalled fork. ATR kinase triggers DNA replication checkpoint signaling in response to replication stress. ssDNA, generated at the stalled replication fork, is coated by RPA, which then recruits ATRIP, the activator of ATR. In response to replication stress, Chk1-binding domain (CKBD) in Claspin is phosphorylated. This is conducted primarily by Cdc7 kinase in cancer cells, while CK1γ1 is predominantly responsible in non-cancer cells (Fig. 2; Yang et al., 2019). This phosphorylation is absolutely required for binding of Chk1 to Claspin, a step prerequisite for checkpoint activation.
ATR and Chk1 kinases cooperatively promote replication checkpoint. Chk1 phosphorylation mediated by Claspin and ATR induces Cdc2/cyclin B1-mediated cell cycle arrest and p53-regulated cellular responses to ssDNA breaks, including DNA repair and even apoptosis when the damage is not properly fixed (Chen, 2016; Lanz et al., 2019; Ronco et al., 2017). Cdc25A, a critical factor that promotes G1/S and G2/M transition, is phosphorylated and targeted for proteasomal degradation as a result of Chk1 activation, leading to the delay of S-phase progression and mitotic entry (Goto et al., 2019; Ronco et al., 2017) (Fig. 2). Activation of Chk1 kinase also induces C-terminal acetylation of p53 (Craig et al., 2007; Shieh et al., 2000; Yogosawa & Yoshida, 2018), which enhances p53 DNA-binding affinity, resulting in upregulation of its downstream target genes (Ou et al., 2005).
Furthermore, one recent study illustrates how inhibition of ATR and Chk1 kinase activities leads to enhanced origin firing. Dual inhibition of ATR and Chk1 kinase activities destabilizes the interactions between Rif1 and phosphatase 1 (PP1), and therefore, PP1 fails to counteract the action of Cdc7 and Cdk, required for initiation, leading to increased origin firing. This is caused by phosphorylation of S2205 of Rif1 by Cdk, and authors suggest that ATR and Chk1 suppress Cdk activity during the unperturbed S phase (Moiseeva et al., 2019) (Fig. 2). Chk1 can also increase replication fork stability in conjunction with poly(ADP-ribose) (PAR) via a C2H2 motif (C8-C6-H8-H) in Chk1 which is conserved in all vertebrates and thus was named as PAR-binding regulatory (PbR) motif (Min et al., 2013). PAR binding to Chk1 also regulates Chk1 kinase activity in an ATR-independent manner, suggesting PAR is also involved in Chk1 activation (Min et al., 2013). Chk1 also affects nucleotide metabolism. RRM1 and RRM2, two subunits of ribonucleotide reductase (RNR) are degraded upon ATR or Chk1 inhibition via increased CDK2 activities (Koppenhafer et al., 2020). At cellular level, as RRM1 and RRM2 are degraded by ATR or Chk1 inhibition, cells enter an apoptotic state due to persistent DNA damages, suggesting a role of ATR and Chk1 in maintenance of nucleotide pools (Koppenhafer et al., 2020). Furthermore, ATR-mediated phosphorylation of Chk1 at Ser-317 and Ser-345 induces its auto-phosphorylation at Ser-296 (Okita et al., 2012). The phosphorylated Ser-296 generates a docking site for 14-3-3γ protein on Chk1 and also enhances interaction between Cdc25A and 14-3-3 protein, promoting the complex formation among Chk1, 14–3-3, and Cdc25A, leading to Cdc25A degradation and preventing mitotic entry (Goto et al., 2014; Kasahara et al., 2010). Chk1 can activate DNA repair pathways by phosphorylating BRCA2 and RAD51 (Bahassi et al., 2008; Enomoto et al., 2009; Ou et al., 2005).
Next, we would like to discuss structural basis on how ATR, Claspin and Chk1 coordinate replication checkpoint. A recent structural analysis demonstrates that dimerized ATR kinase forms a complex with ATR-interacting proteins (ATRIP) (Rao et al., 2018). ATR is composed of N-terminal heat repeats (N-HEAT; residues 1–1383), FAT (FRAP, ATM, TRRAP) domain, a kinase domain (KD), and a C-terminal short fragment referred to as FATC. The structure of ATR–ATRIP complex with a short peptide of Chk1 (residues 343–352) has also been determined. The short Chk1 fragment is modeled into the KD polypeptide and shown to pack against the structure formed by FATC, catalytic loop, and activation loop. Thus, Ser-345 in Chk1 is right in front of the catalytic site of ATR and can be efficiently phosphorylated (Rao et al., 2018). A study on the functional activities of Chk1 phosphorylation sites showed that defective Ser-345 phosphorylation of Chk1 exhibits impaired replication checkpoint and aberrant mitosis, resulting in failure to be localized in the cytoplasm; therefore, Ser-345 phosphorylation is critical for replication checkpoint activation, mitotic progression, and cytoplasmic localization (Niida et al., 2007). On the other hand, Ser-317, another crucial residue for phosphorylation of Chk1, affects replication checkpoint and chromatin binding abilities of Chk1 but not mitotic progression and cytoplasmic localization (Niida et al., 2007), suggesting differential regulation exerted by distinct phosphorylation sites.
Another recent structural study illustrates that Chk1 can recognize phosphorylated Claspin with its Lys-54, Arg-129, Thr-153, and Arg-162 within the kinase domain (Chk1-KD, residues 1–270). These amino acid residues fit to the phosphorylated Ser-945 of Claspin and contribute to the proper interaction between Chk1 and Claspin (Day et al., 2021). It further shows that Chk1-KD can still interact with one of its substrates, Cdc25C, in the presence of bound Claspin, as indicated by fluorescent polarization (FP) assay and NADH-coupled ATPase experiments. These results suggest that Claspin–Chk1 interactions do not have conspicuous impacts on Chk1 kinase activity and that Claspin merely acts as a mediator protein to recruit Chk1 for subsequent replication checkpoint signaling (Day et al., 2021).
Crosstalk between the replication stress checkpoint and general biological stresses
It has been reported that ATR/Claspin/Chk1 or Mec1(Rad3)/Mrc1/Rad53(Cds1) can be activated by various biological stresses (Duch et al., 2013, 2018; Tuul et al., 2013). In budding yeast, heat shock, osmotic stress, hydrogen peroxide (H2O2), and nutrient deprivation are shown to induce replication inhibition, in a manner independent of Mec1 and Rad53 (Bennett & Clarke, 2006; Duch et al., 2013, 2018; Tuul et al., 2013) (Fig. 4A). However, it depends on Mrc1, and critical phosphorylation events are identified that occur in response to these stresses. A comprehensive kinase screening reveals that multiple stress-activated protein kinases (SAPKs) are capable of phosphorylating the specific N-terminal target residues in response to specific cellular stress (Duch et al., 2018). For example, Hog1 interacts with and phosphorylates Mrc1 upon osmotic stress, coordinating replication program with replication stress induced by transcription–replication collision (Duch et al., 2013, 2018). The hog1 mutants fail to phosphorylate Mrc1 and does not slow down S phase, exhibiting DNA damages. Also, phosphorylated Mrc1 induced by Hog1 promotes Cdc45 unloading and reduces replication fork rate (Duch et al., 2013). Likewise, heat stress, oxidative stress or glucose deprivation induces Mpk1, Psk1 and Snf1, respectively, which phosphorylate the critical N-terminal target residues of Mrc1 (Duch et al., 2018). In mammalian cells, Claspin–Chk1 is activated by Unfolded Protein Response (UPR) induced by stresses such as hypoxia to slow down replication fork and reduce origin firing (Fig. 4B). The inhibition of DNA synthesis depends on UPR effector PERK, and is associated with phosphorylation of Claspin (Cabrera et al., 2017). On the other hand, hypoxia induced Senataxin, an RNA–DNA hybrid helicase, in a PERK-dependent manner. Senataxin decreases the numbers of DNA–DNA hybrids and protects cells from DNA damages (Ramachandran et al., 2021).
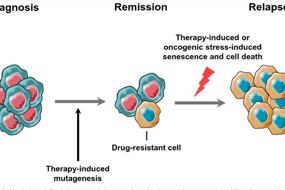
Fig. 4
Claspin/Mrc1 coordinates replication checkpoint signaling in response to various biological stresses. A Different cellular stresses can induce stress-specific kinases, which phosphorylate Mrc1, triggering replication checkpoint signaling in budding yeast. Hog1, Mpk1, Snf1, and Psk1 in yeast phosphorylate the N-terminal segment of Mrc1 in response to osmotic stress, heat shock, glucose deprivation, and oxidative stress, respectively (Duch et al., 2013, 2018). This was termed “Mrc1 transcription–replication safeguard mechanism” and was proposed to serve for maintaining genomic integrity in response to various cellular stresses. B In mammalian cells, hypoxia induces unfolded protein response through PERK, and this would lead to Claspin phosphorylation and Chk1 activation in a manner dependent on PERK. Hypoxia induces transcription-replication collision, which results in formation of RNA–DNA hybrids (R-loop). This also would lead to replication stress-induced Claspin–Chk1 activation. Hypoxia also induces Senataxin (RNA–DNA hybrid helicase) through PERK-ATF4, which antagonizes the formation of RNA–DNA hybrids and reduces the replication stress
Full size imageWe observe that Chk1 is activated by a spectrum of cellular stresses, including heat, osmotic stress, arsenate, oxidative stress, hypoxic stress, glucose shock and so forth, in mammalian cells, and this activation relies on Claspin (Yang et al., unpublished data). Some of these cellular stresses appear to directly induce replication stress, whereas others may activate Claspin–Chk1 through pathways distinct from replication stress.
In conclusion, Mrc1/Claspin coordinates biological stress signals as a general mediator which may or may not activate the downstream effector kinase (Rad53 or Chk1). Currently, upstream sensor and downstream effector kinases of these stress-induced replication checkpoint are being examined in more detail.
The biological significance of the ATR-Claspin-Chk1 axis in oncogenesis and clinical phenotypes in cancer patients
Genome instability is a major driving force that contributes to cancer development and it would be crucial to understand the molecular basis of genome instability to develop novel therapeutic strategies. Therefore, major efforts have been made on studies of mechanistic links between the impaired replication checkpoint control and genome instability. Claspin plays a central role as a mediator between ATR and Chk1, maintaining replication fork stability and safeguarding the genome (Faustrup et al., 2009; Goto et al., 2019; Ito et al., 2018; Kim et al., 2008; Martin et al., 2015; McGarry et al., 2016; Scorah & McGowan, 2009; Sørensen et al., 2003; Yang et al., 2019; Yuan et al., 2014). The ATR-Claspin-Chk1 pathway ensures genome integrity and potentially prevents oncogenesis by several proposed mechanisms, including inhibition of mitotic progression with incompletely duplicated genome, suppression of initiation at dormant origins under replication stress (e.g., HU treatment), and regulation of dNTP pool (Lecona & Fernandez-Capetillo, 2018; Oakes et al., 2014).
In the following section, we would like to discuss clinical correlation between the ATR-Claspin-Chk1 axis and cancer formation. ATR was shown to be upregulated in adenoid cystic carcinoma (ACC), mediated by MYB, a transcription factor noted to be activated in ACC, and this may affect ATR-Claspin-Chk1 signaling (Andersson et al., 2020). However, general roles of Claspin in oncogenesis are still controversial, as it could either support or inhibit cancer cell growth depending on circumstances (Azenha et al., 2017; Bianco et al., 2019; Cai et al., 2021; Kobayashi et al., 2019, 2020; Wang et al., 2017; Yuan et al., 2014). For instance, a recent report shows that Claspin and Tim are overexpressed in primary lung, colorectal and breast cancer specimens and that Claspin together with Tim protein can enhance cell proliferation in untransformed fibroblasts and HCT116 cells, a colorectal carcinoma cell line (Bianco et al., 2019). This report further shows that reduction of Claspin and Tim expression decelerates replication fork progression in HCT116 cells and these two proteins promote the tolerance of oncogene-induced replication stress where an oncogenic form of Ras protein is introduced. Overall, higher expression levels of Claspin and Tim are positively associated in some primary cancer cells and cancer cell lines, and protects cancer cells from oncogene-induced replication stress in a checkpoint-independent manner (Bianco et al., 2019). On the other hand, Claspin may also possess tumor suppressive functions. For example, the expression level of Claspin can be stabilized by USP20, a peptidase for ubiquitin that targets Claspin for degradation, in gastric cancer (GC) cells and a recent study shows that USP20 expression is positively correlated with expression levels of Claspin and that a lower expression level of Claspin is intimately linked to poorer survival and prognosis in GC patients. Indeed, Claspin is one of the major substrates targeted by USP20 (Yuan et al., 2014) and both can suppress tumorigenesis (Wang et al., 2017). Hence, the dual functions of Claspin, both positive and negative, in carcinogenesis require further investigation. Furthermore, Chk1 overexpression has also been noted in several malignant cancers, including breast cancer, T-cell acute lymphoblastic leukemia (T-ALL), and neuroblastoma (Ando et al., 2019; Sarmento et al., 2015; Wu et al., 2019). For instance, Chk1 is overexpressed at mRNA level both in T-ALL cell lines and clinical specimens of T-ALL patients (Sarmento et al., 2015). Chk1 inhibition in T-ALL cell lines causes premature occurrence of DNA replication and induces significantly higher levels of DNA damage and cancer cell death. In T-ALL xenograft models, Chk1 inhibition also retards tumor formation.
Claspin has also been linked to oncovirus-associated cancer formation (Benevolo et al., 2012; Koganti et al., 2014, 2020; Spardy et al., 2009). It has been shown that several oncogenic viruses interfere with the ATR-Claspin-Chk1 pathway. Oncogenic Epstein–Barr virus (EBV) activates STAT3 in B lymphocytes, a transcriptional factor that induces a caspase cascade involving caspase 9 and caspase 7. Caspase 7 then targets Claspin for proteasomal degradation and inhibits Chk1 phosphorylation at Ser-345 (Koganti et al., 2020). Disrupted Claspin–Chk1 pathway permits EBV-infected cells to continuously proliferate regardless of oncovirus-induced replication stress and DNA lesions, leading to active viral replication and tumorigenesis.
To conclude, acquired mutations and anomalous expression in any component of the ATR-Claspin-Chk1 axis or disruption of its operation can perturb replication stress signaling pathway. The level of Claspin can be high or low in different cancer cell lines, and thus, the high level of Claspin expression could trigger oncogenesis in some cases and in others suppress oncogenesis (Bianco et al., 2019; Cai et al., 2021; Kobayashi et al., 2019, 2020; Wang et al., 2017; Yuan et al., 2014). This suggests the potential of the ATR-Claspin-Chk1 axis as an attractive therapeutic target for cancer treatment, but the strategy could be different depending on the nature of the cancer cells.
Targeting the ATR-Claspin-Chk1 axis and its utilization as biomarkers for cancer therapeutics
Manipulation of the ATR-Claspin-Chk1 replication checkpoint pathway could be a potential target of novel cancer treatment strategy (Azenha et al., 2017). Cancer cells possess accumulating genetic alterations due to the defective DDR responses (Dieltein et al., 2014; Burgess et al., 2020). The aberrations of replication checkpoint and the subsequent impairment of the DDR signaling pathways would permit cancer cells to continue to progress through the cell cycle in the presence of replication stress. Frequent upregulation of components for the ATR-Claspin-Chk1 axis in clinical cancer samples leads to the proposal that cancer cells are more heavily dependent on the ATR-Claspin-Chk1 pathway for survival through replication stress compared to normal cells. Thus, targeting this pathway and the associated DDR in cancer cells may render them more susceptible to replication impediments, thereby inducing cancer cell death (Bianco et al., 2019; Cai et al., 2021; Choi et al., 2014; Gilad et al., 2010; Kobayashi et al., 2019, 2020; Tsimaratou et al., 2007). Accordingly, inhibition of the ATR-Claspin-Chk1 axis in combination with added replication stress has been exploited to suppress cancer cell growth and now is under clinical trials for several cancer treatment regimen (Boudny & Trbusek, 2020; Gralewska et al., 2020; Sanjiv et al., 2016; Barnieh et al., 2021; Dent, 2019) (Tables 1, 2, 3, 4).
Table 1 ATR inhibitors under clinical trials



For instance, VE-821, an efficacious ATR inhibitor, strongly suppresses ATR signaling exemplified by significantly downregulated Chk1 phosphorylation at Ser345, a marker for checkpoint activation and a critical residue for subsequent replication checkpoint activation (Huang & Zhou, 2020). The drug increases γH2AX signals and decreases Rad51 foci in primary and cultured pancreatic cancer cells (Prevo et al., 2012), suggesting reduced DNA repair in the presence of the ATR inhibitor. Moreover, AZD7762, a Chk1/2 inhibitor, greatly sensitizes cancer cells to gemcitabine, an inhibitor of DNA synthesis, in urothelial cancer cell lines (UCCs). In MCF-10A (human normal breast epithelial cell cells) and B16-F10 (melanoma cells), AZD7762 in combination with ionizing radiation results in abscopal tumor response through increased micronuclei formation and immune activation signaling (Chao et al., 2020; Prevo et al., 2012). AZD7762 also sensitizes urothelial carcinoma cells to gemcitabine (ionizing radiation mimetics) by inhibiting DNA repair and disturbing checkpoints, supporting the combination of gemcitabine with Chk1 inhibition as a promising urothelial cancer therapy (Isono et al., 2017). AZD7762 also induces synthetic lethality in combination with ATR inhibition (VE-821) in U2OS osteosarcoma cancer cell line, through replication fork arrest, ssDNA accumulation and replication collapse (Sanjiv et al., 2016).
As with other factors for replication checkpoint, Claspin is more frequently upregulated in cancer tissues. Although no Claspin inhibitors have been developed for clinical trials so far, Claspin has been utilized as a proliferation biomarker since its anomalous expression is associated with oncogenic progression, resistance to cancer chemotherapies, and metastases (Choi et al., 2014; Tsimaratou et al., 2007). Upregulation of Claspin expression either at the mRNA or at the protein level has been clinicopathologically shown in certain types of cancer, including, GC, renal cell carcinoma, colorectal carcinoma, prostate cancer, lung cancer, brain cancer, and so on (Bianco et al., 2019; Cai et al., 2021; Choi et al., 2014; Kobayashi et al., 2019, 2020; Tsimaratou et al., 2007). For instance, in prostate cancer, a recent report shows that Claspin overexpression is associated with tumor progression, more aggressive and metastatic nature of the tumor, and relatively poor survival rate in patients, indicating an oncogenic role of Claspin (Kobayashi et al., 2019). Elevated levels of Claspin mRNA and protein in clinical samples may suggest the therapeutic as well as diagnostic and prognostic potential of Claspin in these types of cancers.
Conclusion
In past decades, a wide range of studies on the ATR-Claspin-Chk1 replication checkpoint pathway has revolutionized our perspective regarding the molecular mechanisms of replication checkpoint in oncogenic signaling and its utilization for novel cancer therapies. However, Claspin is differentially involved in the regulation of replication and checkpoint depending on the cell types, tissue, and cancer types in clinical settings. For example, Cdc7-mediated phosphorylation of Claspin at CKBD may be more specific to cancer cells, and in normal cells, CK1γ1 would play a more significant role. In fission yeast, Mrc1 exerts brake for initiation, and Cdc7(Hsk1)-mediated phosphorylation releases this break for initiation. Tumor suppressive role of Claspin in some cancer cells may reflect a potential negative role of Claspin for initiation. Thus, development of anti-Claspin agents for cancer therapy would require further careful investigation.
Future perspectives
Recent studies on Claspin described above have deepened our understanding of how Claspin facilitates initiation and replication fork progression, and maintains genome integrity in the presence of replication stress and how its dysfunction potentially gives rise to oncogenesis. Claspin/Mrc1 plays positive roles in replication initiation and fork progression during normal course of DNA replication, and it would negatively regulate replication upon replication stress and also potentially the initiation stage. Studies on budding yeast Mrc1 indicate that it may act as a brake for replication initiation for fine tuning of initiation timing (Matsumoto et al., 2017). The mechanisms of this negative regulation by Clapsin/Mrc1 of both DNA chain elongation and initiation need to be clarified. Indeed, a recent report indicates Rad53 (effector kinase)-mediated phosphorylation of Mrc1 in response to replication stress inhibits its fork progression activity (McClure & Diffley, 2021).
Besides, roles of Claspin in various cell types and during development have not been investigated. Knockout of Claspin is embryonic lethal at E12.5, suggesting its crucial roles for early development (Yang et al., 2016). However, tissue-specific KO of Claspin may lead to different phenotypes. Claspin’s roles as negative or positive regulator of DNA replication in various organs or tissues need to be carefully evaluated.
Moreover, there are some reports that Claspin–Chk1 is activated by other cellular stresses. In budding yeast, osmotic shock activates Mrc1 through phosphorylation by Hog1, resulting in the inhibition of DNA replication (Duch et al., 2018). Effects of various biological stresses, such as heat, nutrition deprivation, hypoxia and so forth, on the Claspin–Chk1 axis need to be evaluated to clarify how these stress pathways may crosstalk with replication checkpoint pathway.
Furthermore, the structural basis of Claspin is still lacking. It has strongly been indicated that Claspin undergoes intramolecular interaction, which may be regulated by Cdc7-mediated phosphorylation (Masai et al., 2017; Yang et al., 2016). Claspin, predicted to harbor large segments of intrinsically disordered polypeptide (IDP), may adopt variable structures depending on its bound partners and covalent modifications. Clarification on structures of Claspin under various conditions will provide important information on its modes of action at the initiation, replication fork progression and replication checkpoint.
Finally, it is also important to clarify the roles of Claspin during oncogenesis of various cancer types. Taken together, these future endeavors will lead to identification of novel diagnostic and prognostic cancer biomarkers as well as effective therapeutic strategies involving Claspin as a target.
References
Andersson, M. K., Mangiapane, G., Nevado, P. T., Tsakaneli, A., Carlsson, T., Corda, G., Nieddu, V., Abrahamian, C., Chayka, O., Rai, L., Wick, M., Kedaigle, A., Stenman, G., & Sala, A. (2020). ATR is a MYB regulated gene and potential therapeutic target in adenoid cystic carcinoma. Oncogenesis. //doi.org/10.1038/s41389-020-0194-3 Article number: 5.
Ando, K., Nakamura, Y., Nagase, H., Nakagawara, A., Koshinaga, T., Wada, S., & Makishima, M. (2019). Co-inhibition of the DNA damage response and CHK1 enhances apoptosis of neuroblastoma cells. International Journal of Molecular Sciences, 20(15), 3700. //doi.org/10.3390/ijms20153700
Andor, N., Maley, C. C., & Ji, H. P. (2017). Genomic instability in cancer: Teetering on the limit of tolerance. Cancer Research, 77(9), 2179–2185. //doi.org/10.1158/0008-5472.CAN-16-1553
Archie, N. T., Rendahl, K. G., Sheikh, T., Cheema, H., Aardalen, K., Embry, M., Ma, S., Moler, E. J., Ni, Z. J., Lopes de MenezesHibner, D. E. B., Gesner, T. G., & Schwartz, G. K. (2007). CHIR-124, a novel potent inhibitor of Chk1, potentiates the cytotoxicity of topoisomerase I poisons in vitro and in vivo. Clinical Cancer Research, 13(2), 591–602. //doi.org/10.1158/1078-0432.CCR-06-1424
Azenha, D., Lopes, M. C., & Martins, T. C. (2017). Claspin functions in cell homeostasis—A link to cancer? DNA Repair, 59, 27–33. //doi.org/10.1016/j.dnarep.2017.09.002
Azenha, D., Lopes, M. C., & Martins, T. C. (2019). Claspin: From replication stress and DNA damage responses to cancer therapy. Advances in Protein Chemistry and Structural Biology, 115, 203–246. //doi.org/10.1016/bs.apcsb.2018.10.007
Bacal, J., Morielcarretero, M., Pardo, B., Barthe, A., Sharma, S., Chabes, A., Lengronne, A., & Pasero, P. (2018). Mrc1 and Rad9 cooperate to regulate initiation and elongation of DNA replication in response to DNA damage. The EMBO Journal, 37(21), e99319. //doi.org/10.15252/embj.201899319
Bahassi, E. M., Ovesen, J. L., Riesenberg, A. L., Bernstein, W. Z., Hasty, P. E., & Stambrook, P. J. (2008). The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene, 27(28), 3977–3985. //doi.org/10.1038/onc.2008.17
Baretić, D., Jenkyn-Bedford, M., Aria, V., Cannone, G., Skehel, M., & Yeeles, J. T. (2020). Cryo-EM structure of the fork protection complex bound to CMG at a replication fork. Molecular Cell, 78(5), 926–940. //doi.org/10.1016/j.molcel.2020.04.012
Barker, H. E., Patel, R., McLaughlin, M., Schick, U., Zaidi, S., Nutting, C. M., Newbold, K. L., & Harrington, K. J. (2016). CHK1 inhibition radiosensitizes head and neck cancers to paclitaxel-based chemoradiotherapy. Molecular Cancer Therapeutics, 15(9), 2042–2054. //doi.org/10.1158/1535-7163.MCT-15-0998
Barnieh, F. M., Loadman, P. M., & Falconer, R. A. (2021). Progress towards a clinically-successful ATR inhibitor for cancer therapy. Current Research in Pharmacology and Drug Discovery. //doi.org/10.1016/j.crphar.2021.100017
Bartkova, J., Rezaei, N., Liontos, M., et al. (2006). Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature, 444, 633–637. //doi.org/10.1038/nature05268
Benevolo, M., Musio, A., Vocaturo, A., Donà, M. G., Rollo, F., Terrenato, I., Carosi, M., Pescatmona, E., Vocaturo, G., & Mottolese, M. (2012). Claspin as a biomarker of human papillomavirus-related high grade lesions of uterine cervix. Journal of Translational Medicine, 10(1), 1–8. //doi.org/10.1186/1479-5876-10-132
Bennett, L. N., & Clarke, P. R. (2006). Regulation of Claspin degradation by the ubiquitin-proteosome pathway during the cell cycle and in response to ATR-dependent checkpoint activation. FEBS Letters, 580(17), 4176–4181. //doi.org/10.1016/j.febslet.2006.06.071
Berens, T. J., & Toczyski, D. P. (2012). Colocalization of Mec1 and Mrc1 is sufficient for Rad53 phosphorylation in vivo. Molecular Biology of the Cell, 23(6), 1058–1067. //doi.org/10.1091/mbc.e11-10-0852
Bianco, J. N., Bergoglio, V., Lin, Y. L., Pillaire, M. J., Schmitz, A. L., Gilhodes, J., Lusque, A., Mazières, J., Lacroix-Triki, M., Roumeliotis, T. I., Choudhary, J., Moreaux, J., Hoffmann, J. S., Tourrière, H., & Pasero, P. (2019). Overexpression of Claspin and Timeless protects cancer cells from replication stress in a checkpoint-independent manner. Nature Communications, 10(1), 1–14. //doi.org/10.1038/s41467-019-08886-8
Blasina, A., Hallin, J., Chen, E., Arango, M. E., Kraynov, E., Register, J., Grant, S., Ninkovic, S., Chen, P., Nichols, T., O’Connor, P., & Anderes, K. (2008). Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Molecular Cancer Therapeutics, 7(8), 2394–2404. //doi.org/10.1158/1535-7163.MCT-07-2391
Boudny, M., & Trbusek, M. (2020). ATR-CHK1 pathway as a therapeutic target for acute and chronic leukemias. Cancer Treatment Reviews. //doi.org/10.1016/j.ctrv.2020.102026
Brooks, K., Oakes, V., Edwards, B., Ranall, M., Leo, P., Pavey, S., Pinder, A., Beamish, H., Mukhopadhyay, P., Lambie, P., & Gabrielli, B. (2013). A potent Chk1 inhibitor is selectively cytotoxic in melanomas with high levels of replicative stress. Oncogene, 32(6), 788–796. //doi.org/10.1038/onc.2012.72
Burgess, J. T., Rose, M., Boucher, D., Plowman, J., Molloy, C., Fisher, M., O’Leary, C., Richard, D., O’Byrne, K. J., & Bolderson, E. (2020). The therapeutic potential of DNA damage repair pathways and genomic stability in lung cancer. Frontiers in Oncology. //doi.org/10.3389/fonc.2020.01256
Cabrera, E., Hernández-Pérez, S., Koundrioukoff, S., et al. (2017). PERK inhibits DNA replication during the unfolded protein response via Claspin and Chk1. Oncogene, 36, 678–686. //doi.org/10.1038/onc.2016.239
Cai, C., Luo, J., Liu, Q., Liu, Z., Zhao, Y., Wu, X., Yuegao, Y., Lei, Y., Lu, J., Wang, Y., Cai, Z., Duan, X., Lei, M., Gu, D., & Liu, Y. (2021). Claspin overexpression promotes tumor progression and predicts poor clinical outcome in prostate cancer. Genetic Testing and Molecular Biomarkers, 25(2), 131–139. //doi.org/10.1089/gtmb.2020.0226
Cazares-Körner, C., Pires, I. M., Swallow, I. D., Grayer, S. C., O’Connor, L. J., Olcina, M. M., Christlieb, M., Conway, S. J., & Hammond, E. M. (2013). CH-01 is a hypoxia-activated prodrug that sensitizes cells to hypoxia/reoxygenation through inhibition of Chk1 and Aurora A. ACS Chemical Biology, 8(7), 1451–1459. //doi.org/10.1021/cb4001537
Chao, H. H., Karagounis, I. V., Thomas, C., François, N. B., Facciabene, A., Koumenis, C., & Maity, A. (2020). Combination of CHEK1/2 inhibition and ionizing radiation results in abscopal tumor response through increased micronuclei formation. Oncogene, 39(22), 4344–4357. //doi.org/10.1038/s41388-020-1300-x
Chen, J. (2016). The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harbor Perspectives in Medicine, 6(3), a026104. //doi.org/10.1101/cshperspect.a026104
Choi, S. H., Yang, H., Lee, S. H., Ki, J. H., Nam, D. H., & Yoo, H. Y. (2014). TopBP1 and Claspin contribute to the radioresistance of lung cancer brain metastases. Molecular Cancer, 13(1), 1–8. //doi.org/10.1186/1476-4598-13-211
Chopra, S. S., Jenney, A., Palmer, A., Niepel, M., Chung, M., Mills, C., et al. (2020). Torin2 exploits replication and checkpoint vulnerabilities to cause death of PI3K-activated triple-negative breast cancer cells. Cell Systems, 10(1), 66–81. //doi.org/10.1016/j.cels.2019.11.001
Craig, A. L., Chrystal, J. A., Fraser, J. A., Sphyris, N., Lin, Y., Harrison, B. J., Scott, M. T., Dornreiter, I., & Hupp, T. R. (2007). The MDM2 ubiquitination signal in the DNA-binding domain of p53 forms a docking site for calcium calmodulin kinase superfamily members. Molecular and Cellular Biology, 27(9), 3542–3555. //doi.org/10.1128/MCB.01595-06
Day, M., Parry-Morris, S., Houghton-Gisby, J., Oliver, A. W., & Pearl, L. H. (2021). Structural basis for recruitment of the CHK1 DNA damage kinase by the CLASPIN scaffold protein. Structure. //doi.org/10.1016/j.str.2021.03.007
Dent, P. (2019). Investigational CHK1 inhibitors in early phase clinical trials for the treatment of cancer. Expert Opinion on Investigational Drugs, 28(12), 1095–1100. //doi.org/10.1080/13543784.2019.1694661
Di Micco, R., Fumagalli, M., Cicalese, A., et al. (2006). Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature, 444, 638–642. //doi.org/10.1038/nature05327
Dietlein, F., Thelen, L., & Reinhardt, H. C. (2014). Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends in Genetics, 30(8), 326–339. //doi.org/10.1016/j.tig.2014.06.003
Dillon, M. T., Barker, H. E., Pedersen, M., Hafsi, H., Bhide, S. A., Newbold, K. L., Nutting, C. M., McLaughlin, M., & Harrington, K. J. (2017). Radiosensitization by the ATR inhibitor AZD6738 through generation of acentric micronuclei. Molecular Cancer Therapeutics, 16(1), 25–34. //doi.org/10.1158/1535-7163.MCT-16-0239
Duch, A., Canal, B., Barroso, S. I., García-Rubio, M., Seisenbacher, G., Aguilera, A., Nadal, E., & Posas, F. (2018). Multiple signaling kinases target Mrc1 to prevent genomic instability triggered by transcription-replication conflicts. Nature Communications, 9(1), 1–14. //doi.org/10.1038/s41467-017-02756-x
Duch, A., Felipe-Abrio, I., Barroso, S., Yaakov, G., Garcia-Rubio, M., Aguilera, A., Nadal, E., & Posas, F. (2013). Coordinated control of replication and transcription by a SAPK protects genomic integrity. Nature, 493(7430), 116–119. //doi.org/10.1038/nature11675
Enomoto, M., Goto, H., Tomono, Y., Kasahara, K., Tsujimura, K., Kiyono, T., & Inagaki, M. (2009). Novel positive feedback loop between Cdk1 and Chk1 in the nucleus during G2/M transition. Journal of Biological Chemistry, 284(49), 34223–34230. //doi.org/10.1074/jbc.C109.051540
Faustrup, H., Bekker-Jensen, S., Bartek, J., Lukas, J., & Mailand, N. (2009). USP7 counteracts SCFβTrCP-but not APCCdh1-mediated proteolysis of Claspin. The Journal of Cell Biology, 184(1), 13–19. //doi.org/10.1083/jcb.200807137
Foote, K. M., Lau, A., & M Nissink, J. W. (2015). Drugging ATR: progress in the development of specific inhibitors for the treatment of cancer. Future Medicinal Chemistry, 7(7), 873–891. //doi.org/10.4155/fmc.15.33
Froget, B., Blaisonneau, J., Lambert, S., & Baldacci, G. (2008). Cleavage of stalled forks by fission yeast Mus81/Eme1 in absence of DNA replication checkpoint. Molecular Biology of the Cell, 19(2), 445–456. //doi.org/10.1091/mbc.e07-07-0728
Gaillard, H., García-Muse, T., & Aguilera, A. (2015). Replication stress and cancer. Nature Reviews Cancer, 15(5), 276–289. //doi.org/10.1038/nrc3916
Gambus, A., Jones, R. C., Sanchez-Diaz, A., Kanemaki, M., Van Deursen, F., Edmondson, R. D., & Labib, K. (2006). GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nature Cell Biology, 8(4), 358–366. //doi.org/10.1038/ncb1382
Gellon, L., Kaushal, S., Cebrián, J., Lahiri, M., Mirkin, S. M., & Freudenreich, C. H. (2019). Mrc1 and Tof1 prevent fragility and instability at long CAG repeats by their fork stabilizing function. Nucleic Acids Research, 47(2), 794–805. //doi.org/10.1093/nar/gky1195
Gilad, O., Nabet, B. Y., Ragland, R. L., Schoppy, D. W., Smith, K. D., Durham, A. C., & Brown, E. J. (2010). Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Research, 70(23), 9693–9702. //doi.org/10.1158/0008-5472.CAN-10-2286
Gorgoulis, V. G., Vassiliou, L. V. F., Karakaidos, P., Zacharatos, P., Kotsinas, A., Liloglou, T., Venere, M., DiTullio, R. A., Kastrinakis, N. G., Levy, B., Kletsas, D., Yoneta, A., & Halazonetis, T. D. (2005). Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature, 434(7035), 907–913. //doi.org/10.1038/nature03485
Goto, H., Kasahara, K., & Inagaki, M. (2014). Novel insights into Chk1 regulation by phosphorylation. Cell Structure and Function. //doi.org/10.1247/csf.14017
Goto, H., Natsume, T., Kanemaki, M. T., Kaito, A., Wang, S., Gabazza, E. C., Inagaki, M., & Mizoguchi, A. (2019). Chk1-mediated Cdc25A degradation as a critical mechanism for normal cell cycle progression. Journal of Cell Science. //doi.org/10.1242/jcs.223123
Gralewska, P., Gajek, A., Marczak, A., & Rogalska, A. (2020). Participation of the ATR/CHK1 pathway in replicative stress targeted therapy of high-grade ovarian cancer. Journal of Hematology & Oncology, 13, 1–16. //doi.org/10.1186/s13045-020-00874-6
Hall, A. B., Newsome, D., Wang, Y., Boucher, D. M., Eustace, B., Gu, Y., Hare, B., Johnson, M. A., Milton, S., Murphy, C. E., Takemoto, D., Tolman, C., Wood, M., Charlton, P., Charrier, J. D., Furey, B., Golec, J., Reaper, P. M., & Pollard, J. R. (2014). Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget, 5(14), 5674. //doi.org/10.18632/oncotarget.2158
Hayano, M., Kanoh, Y., Matsumoto, S., Kakusho, N., & Masai, H. (2011). Mrc1 marks early-firing origins and coordinates timing and efficiency of initiation in fission yeast. MolEcular and Cellular Biology, 31, 2380–2389. //doi.org/10.1128/MCB.01239-10
Huang, R. X., & Zhou, P. K. (2020). DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduction and Targeted Therapy, 5(1), 1–27. //doi.org/10.1038/s41392-020-0150-x
Isono, M., Hoffmann, M. J., Pinkerneil, M., Sato, A., Michaelis, M., Cinatl, J., Niegisch, G., & Schulz, W. A. (2017). Checkpoint kinase inhibitor AZD7762 strongly sensitises urothelial carcinoma cells to gemcitabine. Journal of Experimental & Clinical Cancer Research, 36(1), 1–12. //doi.org/10.1186/s13046-016-0473-1
Ito, F., Yoshimoto, C., Yamada, Y., Sudo, T., & Kobayashi, H. (2018). The HNF-1β—USP28—Claspin pathway upregulates DNA damage-induced Chk1 activation in ovarian clear cell carcinoma. Oncotarget, 9(25), 17512. //doi.org/10.18632/oncotarget.24776
Kasahara, K., Goto, H., Enomoto, M., Tomono, Y., Kiyono, T., & Inagaki, M. (2010). 14-3-3γ mediates Cdc25A proteolysis to block premature mitotic entry after DNA damage. The EMBO Journal, 29(16), 2802–2812. //doi.org/10.1038/emboj.2010.157
Kemp, M. G., Akan, Z., Yilmaz, S., Grillo, M., Smith-Roe, S. L., Kang, T. H., Cordeiro-Stone, M., Kaufmann, W. K., Abraham, R. T., Sancar, A., & Ünsal-Kaçmaz, K. (2010). Tipin-replication protein A interaction mediates Chk1 phosphorylation by ATR in response to genotoxic stress. Journal of Biological Chemistry, 285(22), 16562–16571. //doi.org/10.1074/jbc.M110.110304
Kim, J. M., Kakusho, N., Yamada, M., Kanoh, Y., Takemoto, N., & Masai, H. (2008). Cdc7 kinase mediates Claspin phosphorylation in DNA replication checkpoint. Oncogene, 27(24), 3475–3482. //doi.org/10.1038/sj.onc.1210994
King, C., Diaz, H., Barnard, D., Barda, D., Clawson, D., Blosser, W., Cox, K., Guo, S., & Marshall, M. (2014). Characterization and preclinical development of LY2603618: A selective and potent Chk1 inhibitor. Investigational New Drugs, 32(2), 213–226. //doi.org/10.1007/s10637-013-0036-7
King, C., Diaz, H. B., McNeely, S., Barnard, D., Dempsey, J., Blosser, W., Beckmann, R., Barda, D., & Marshall, M. S. (2015). LY2606368 causes replication catastrophe and antitumor effects through CHK1-dependent mechanisms. Molecular Cancer Therapeutics, 14(9), 2004–2013. //doi.org/10.1158/1535-7163.MCT-14-1037
Klein, C. A. (2020). Cancer progression and the invisible phase of metastatic colonization. Nature Reviews Cancer, 20(11), 681–694. //doi.org/10.1038/s41568-020-00300-6
Kobayashi, G., Sentani, K., Babasaki, T., Sekino, Y., Shigematsu, Y., Hayashi, T., Oue, N., Teishima, J., Matsubara, A., Sasaki, N., & Yasui, W. (2020). Claspin overexpression is associated with high-grade histology and poor prognosis in renal cell carcinoma. Cancer Science, 111(3), 1020. //doi.org/10.1111/cas.14299
Kobayashi, G., Sentani, K., Hattori, T., Yamamoto, Y., Imai, T., Sakamoto, N., Kuraoka, K., Oue, N., Sasaki, N., Taniyama, K., & Yasui, W. (2019). Clinicopathological significance of claspin overexpression and its association with spheroid formation in gastric cancer. Human Pathology, 84, 8–17. //doi.org/10.1016/j.humpath.2018.09.001
Koganti, S., Burgula, S., & Bhaduri-McIntosh, S. (2020). STAT3 activates the anti-apoptotic form of caspase 9 in oncovirus-infected B lymphocytes. Virology, 540, 160–164. //doi.org/10.1016/j.virol.2019.11.017
Koganti, S., Hui-Yuen, J., McAllister, S., Gardner, B., Grasser, F., Palendira, U., Tangye, S. G., Freeman, A. F., & Bhaduri-McIntosh, S. (2014). STAT3 interrupts ATR-Chk1 signaling to allow oncovirus-mediated cell proliferation. Proceedings of the National Academy of Sciences of the United States of America, 111, 4946–4951. //doi.org/10.1073/pnas.1400683111
Koppenhafer, S. L., Goss, K. L., Terry, W. W., & Gordon, D. J. (2020). Inhibition of the ATR–CHK1 pathway in Ewing Sarcoma cells causes DNA damage and apoptosis via the CDK2-mediated degradation of RRM2. Molecular Cancer Research, 18(1), 91–104. //doi.org/10.1158/1541-7786.MCR-19-0585
Lanz, M. C., Dibitetto, D., & Smolka, M. B. (2019). DNA damage kinase signaling: checkpoint and repair at 30 years. The EMBO Journal, 38(18), e101801. //doi.org/10.15252/embj.2019101801
Lara, P. N., Mack, P. C., Synold, T., Frankel, P., Longmate, J., Gumerlock, P. H., Doroshow, J. H., & Gandara, D. R. (2005). The cyclin-dependent kinase inhibitor UCN-01 plus cisplatin in advanced solid tumors: a California cancer consortium phase I pharmacokinetic and molecular correlative trial. Clinical Cancer Research, 11(12), 4444–4450. //doi.org/10.1158/1078-0432.CCR-04-2602
Lecona, E., & Fernandez-Capetillo, O. (2018). Targeting ATR in cancer. Nature Reviews Cancer, 18(9), 586–595. //doi.org/10.1038/s41568-018-0034-3
Lee, J. H., Choy, M. L., Ngo, L., Venta-Perez, G., & Marks, P. A. (2011). Role of checkpoint kinase 1 (Chk1) in the mechanisms of resistance to histone deacetylase inhibitors. Proceedings of the National Academy of Sciences, 108(49), 19629–19634. //doi.org/10.1073/pnas.1117544108
Lee, T. H., Choi, J. Y., Park, J. M., & Kang, T. H. (2020). Posttranscriptional control of the replication stress response via TTP-mediated Claspin mRNA stabilization. Oncogene, 39(16), 3245–3257. //doi.org/10.1038/s41388-020-1220-9
Leman, A. R., Noguchi, C., Lee, C. Y., & Noguchi, E. (2010). Human timeless and Tipin stabilize replication forks and facilitate sister-chromatid cohesion. Journal of Cell Science, 123(5), 660–670. //doi.org/10.1242/jcs.057984
Leman, A. R., & Noguchi, E. (2012). Local and global functions of Timeless and Tipin in replication fork protection. Cell Cycle, 11(21), 3945–3955. //doi.org/10.4161/cc.21989
Lewis, J. S., Spenkelink, L. M., Schauer, G. D., Hill, F. R., Georgescu, R. E., O’Donnell, M. E., & van Oijen, A. M. (2017). Single-molecule visualization of Saccharomyces cerevisiae leading-strand synthesis reveals dynamic interaction between MTC and the replisome. Proceedings of the National Academy of Sciences, 114(40), 10630–10635. //doi.org/10.1073/pnas.1711291114
Li, D. Q., Ohshiro, K., Khan, M. N., & Kumar, R. (2010). Requirement of MTA1 in ATR-mediated DNA damage checkpoint function. Journal of Biological Chemistry, 285(26), 19802–19812. //doi.org/10.1074/jbc.M109.085258
Liu, G., Chen, X., Gao, Y., Lewis, T., Barthelemy, J., & Leffak, M. (2012a). Altered replication in human cells promotes DMPK (CTG) n·(CAG) n repeat instability. Molecular and Cellular Biology, 32(9), 1618–1632. //doi.org/10.1128/MCB.06727-11
Liu, S., Shiotani, B., Lahiri, M., Maréchal, A., Tse, A., Leung, C. C. Y., Glover, J. N. M., Yang, X. H., & Zou, L. (2011). ATR autophosphorylation as a molecular switch for checkpoint activation. Molecular Cell, 43(2), 192–202. //doi.org/10.1016/j.molcel.2011.06.019
Liu, Z., Zhang, B., Liu, K., Ding, Z., & Hu, X. (2012b). Schisandrin B attenuates cancer invasion and metastasis via inhibiting epithelial-mesenchymal transition. PLoS One, 7(7), e40480. //doi.org/10.1371/journal.pone.0040480
Martin, Y., Cabrera, E., Amoedo, H., Hernandez-Perez, S., Dominguez-Kelly, R., & Freire, R. (2015). USP29 controls the stability of checkpoint adaptor Claspin by deubiquitination. Oncogene, 34(8), 1058–1063. //doi.org/10.1038/onc.2014.38
Masai, H., Yang, C. C., & Matsumoto, S. (2017). Mrc1/Claspin: A new role for regulation of origin firing. Current Genetics, 63(5), 813–818. //doi.org/10.1007/s00294-017-0690-y
Matsumoto, S., Hayano, M., Kanoh, Y., & Masai, H. (2011). Multiple pathways can bypass the essential role of fission yeast Hsk1 kinase in DNA replication initiation. Journal of Cell Biology, 195(3), 387–401. //doi.org/10.1083/jcb.201107025
Matsumoto, S., Kanoh, Y., Shimmoto, M., Hayano, M., Ueda, K., Fukatsu, R., Kakusho, N., & Masai, H. (2017). Checkpoint-independent regulation of origin firing by Mrc1 through interaction with Hsk1 kinase. Molecular and Cellular Biology. //doi.org/10.1128/MCB.00355-16
Matsumoto, S., Shimmoto, M., Kakusho, N., Yokoyama, M., Kanoh, Y., Hayano, M., Russell, P., & Masai, H. (2010). Hsk1 kinase and Cdc45 regulate replication stress-induced checkpoint responses in fission yeast. Cell Cycle, 9(23), 4627–4637. //doi.org/10.4161/cc.9.23.13937
Matthews, D. J., Yakes, M., Chen, J., Tadano, M., Bornheim, L., Clary, D. O., Tai, A., Wagner, J. M., Miller, N., Kim, Y. D., Robertson, S., Murray, L., & Karnitz, L. M. (2007). Pharmacological abrogation of S-phase checkpoint enhances the anti-tumor activity of gemcitabine in vivo. Cell Cycle, 6(1), 104–110. //doi.org/10.4161/cc.6.1.3699
McClure, A. W., & Diffley, J. F. (2021). Rad53 checkpoint kinase regulation of DNA replication fork rate via Mrc1 phosphorylation. eLife, 10, e69726. //doi.org/10.7554/eLife.69726
McGarry, E., Gaboriau, D., Rainey, M. D., Restuccia, U., Bachi, A., & Santocanale, C. (2016). The deubiquitinase USP9X maintains DNA replication fork stability and DNA damage checkpoint responses by regulating CLASPIN during S-phase. Cancer Research, 76(8), 2384–2393. //doi.org/10.1158/0008-5472.CAN-15-2890
Min, W., Bruhn, C., Grigaravicius, P., Zhou, Z. W., Li, F., Krüger, A., Siddeek, B., Greulich, K. O., Popp, O., Meisezahl, C., CalkhovenBürkle, C. F. A., Xu, X., & Wang, Z. Q. (2013). Poly (ADP-ribose) binding to Chk1 at stalled replication forks is required for S-phase checkpoint activation. Nature Communications, 4(1), 1–14. //doi.org/10.1038/ncomms3993
Mine, N., Yamamoto, S., Saito, N., Yamazaki, S., Suda, C., Ishigaki, M., Kufe, D. W., Von Hoff, D. D., & Kawabe, T. (2011). CBP501-calmodulin binding contributes to sensitizing tumor cells to cisplatin and bleomycin. Molecular Cancer Therapeutics, 10(10), 1929–1938. //doi.org/10.1158/1535-7163.MCT-10-1139
Moiseeva, T. N., Yin, Y., Calderon, M. J., Qian, C., Schamus-Haynes, S., Sugitani, N., Osmanbeyoglu, H. U., Rothenberg, E., Watkins, S. C., & Bakkenist, C. J. (2019). An ATR and CHK1 kinase signaling mechanism that limits origin firing during unperturbed DNA replication. Proceedings of the National Academy of Sciences, 116(27), 13374–13383. //doi.org/10.1073/pnas.1903418116
Niida, H., Katsuno, Y., Banerjee, B., Hande, M. P., & Nakanishi, M. (2007). Specific role of Chk1 phosphorylations in cell survival and checkpoint activation. Molecular and Cellular Biology, 27(7), 2572–2581. //doi.org/10.1128/MCB.01611-06
Noguchi, E., Noguchi, C., Du, L. L., & Russell, P. (2003). Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Molecular and Cellular Biology, 23(21), 7861–7874. //doi.org/10.1128/MCB.23.21.7861-7874.2003
Oakes, V., Wang, W., Harrington, B., Lee, W. J., Beamish, H., Chia, K. M., Pinder, A., Goto, H., Inagaki, M., Pavey, H. S., & Gabrielli, B. (2014). Cyclin A/Cdk2 regulates Cdh1 and claspin during late S/G2 phase of the cell cycle. Cell Cycle, 13(20), 3302–3311. //doi.org/10.4161/15384101.2014.949111
Okita, N., Minato, S., Ohmi, E., Tanuma, S. I., & Higami, Y. (2012). DNA damage-induced CHK1 autophosphorylation at Ser296 is regulated by an intramolecular mechanism. FEBS Letters, 586(22), 3974–3979. //doi.org/10.1016/j.febslet.2012.09.048
Ou, Y. H., Chung, P. H., Sun, T. P., & Shieh, S. Y. (2005). p53 C-terminal phosphorylation by CHK1 and CHK2 participates in the regulation of DNA-damage-induced C-terminal acetylation. Molecular Biology of the Cell, 16(4), 1684–1695. //doi.org/10.1091/mbc.e04-08-0689
Patties, I., Kallendrusch, S., Böhme, L., Kendzia, E., Oppermann, H., Gaunitz, F., Kortmann, R. F., & Glasow, A. (2019). The Chk1 inhibitor SAR-020106 sensitizes human glioblastoma cells to irradiation, to temozolomide, and to decitabine treatment. Journal of Experimental & Clinical Cancer Research, 38(1), 1–16. //doi.org/10.1186/s13046-019-1434-2
Peasland, A., Wang, L. Z., Rowling, E., Kyle, S., Chen, T., Hopkins, A., Cliby, W. A., Sarkaria, J., Beale, G., Edmondson, R. J., & Curtin, N. J. (2011). Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. British Journal of Cancer, 105(3), 372–381. //doi.org/10.1038/bjc.2011.243
Petermann, E., Helleday, T., & Caldecott, K. W. (2008). Claspin promotes normal replication fork rates in human cells. Molecular Biology of the Cell, 19(6), 2373–2378. //doi.org/10.1091/mbc.e07-10-1035
Prevo, R., Fokas, E., Reaper, P. M., Charlton, P. A., Pollard, J. R., McKenna, W. G., Musche, R. J., & Brunner, T. B. (2012). The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biology & Therapy, 13(11), 1072–1081. //doi.org/10.4161/cbt.21093
Qiu, Z., Oleinick, N. L., & Zhang, J. (2018). ATR/CHK1 inhibitors and cancer therapy. Radiotherapy and Oncology, 126(3), 450–464. //doi.org/10.1016/j.radonc.2017.09.043
Rageul, J., Park, J. J., Zeng, P. P., et al. (2020). SDE2 integrates into the TIMELESS-TIPIN complex to protect stalled replication forks. Nature Communications, 11, 5495. //doi.org/10.1038/s41467-020-19162-5
Rainey, M., Harhen, B., Wang, G. N., Murphy, P., & Santocanale, C. (2013). Cdc7-dependent and-independent phosphorylation of Claspin in the induction of the DNA replication checkpoint. Cell Cycle, 12(10), 1560–1568. //doi.org/10.4161/cc.24675
Ramachandran, S., Ma, T. S., Griffin, J., et al. (2021). Hypoxia-induced SETX links replication stress with the unfolded protein response. Nature Communications, 12, 3686. //doi.org/10.1038/s41467-021-24066-z
Rao, Q., Liu, M., Tian, Y., Wu, Z., Hao, Y., Song, L., Qin, Z., Ding, C., Wang, H. W., Wang, J., & Xu, Y. (2018). Cryo-EM structure of human ATR-ATRIP complex. Cell Research, 28(2), 143–156. //doi.org/10.1038/cr.2017.158
Riesterer, O., Matsumoto, F., Wang, L., Pickett, J., Molkentine, D., Giri, U., Milas, L., & Raju, U. (2011). A novel Chk inhibitor, XL-844, increases human cancer cell radiosensitivity through promotion of mitotic catastrophe. Investigational New Drugs, 29(3), 514–522. //doi.org/10.1007/s10637-009-9361-2
Ronco, C., Martin, A. R., Demange, L., & Benhida, R. (2017). ATM, ATR, CHK1, CHK2 and WEE1 inhibitors in cancer and cancer stem cells. MedChemComm, 8(2), 295–319. //doi.org/10.1039/c6md00439c
Sakakibara, K., Sato, T., Kufe, D. W., VonHoff, D. D., & Kawabe, T. (2017). CBP501 induces immunogenic tumor cell death and CD8 T cell infiltration into tumors in combination with platinum, and increases the efficacy of immune checkpoint inhibitors against tumors in mice. Oncotarget, 8(45), 78277. //doi.org/10.18632/oncotarget.20968
Sanjiv, K., Hagenkort, A., Calderón-Montaño, J. M., Koolmeister, T., Reaper, P. M., Mortusewicz, O., Jacques, S. A., Kuiper, R. V., Schultz, N., Scobie, M., Charlton, P. A., Pollard, J. R., Berglund, U. W., Altun, M., & Helleday, T. (2016). Cancer-specific synthetic lethality between ATR and CHK1 kinase activities. Cell Reports, 14(2), 298–309. //doi.org/10.1016/j.celrep.2016.12.031
Sarmento, L. M., Póvoa, V., Nascimento, R., Real, G., Antunes, I., Martins, L. R., Moita, C., Alves, P. M., Abecasis, M., Moita, L. F., Parkhouse, R. M. E., Meijerink, J. P. P., & Barata, J. T. (2015). CHK1 overexpression in T-cell acute lymphoblastic leukemia is essential for proliferation and survival by preventing excessive replication stress. Oncogene, 34(23), 2978–2990. //doi.org/10.1038/onc.2014.248
Scorah, J., & McGowan, C. H. (2009). Claspin and Chk1 regulate replication fork stability by different mechanisms. Cell Cycle, 8(7), 1036–1043. //doi.org/10.4161/cc.8.7.8040
Shieh, S. Y., Ahn, J., Tamai, K., Taya, Y., & Prives, C. (2000). The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes & Development, 14(3), 289–300. //doi.org/10.1101/gad.14.3.289
Smits, V. A., Cabrera, E., Freire, R., & Gillespie, D. A. (2019). Claspin–checkpoint adaptor and DNA replication factor. The FEBS Journal, 286(3), 441–455. //doi.org/10.1111/febs.14594
Sørensen, C. S., Syljuåsen, R. G., Falck, J., Schroeder, T., Rönnstrand, L., Khanna, K. K., Zhou, B. B., Bartek, J., & Lukas, J. (2003). Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell, 3(3), 247–258. //doi.org/10.1016/S1535-6108(03)00048-5
Spardy, N., Covella, K., Cha, E., Hoskins, E. E., Wells, S. I., Duensing, A., & Duensing, S. (2009). Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Research, 69(17), 7022–7029. //doi.org/10.1158/0008-5472.CAN-09-0925
Suzuki, M., Yamamori, T., Bo, T., Sakai, Y., & Inanami, O. (2017). MK-8776, a novel Chk1 inhibitor, exhibits an improved radiosensitizing effect compared to UCN-01 by exacerbating radiation-induced aberrant mitosis. Translational Oncology, 10(4), 491–500. //doi.org/10.1016/j.tranon.2017.04.002
Técher, H., Koundrioukoff, S., Nicolas, A., & Debatisse, M. (2017). The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nature Reviews Genetics, 18(9), 535–550. //doi.org/10.1038/nrg.2017.46
Tercero, J. A., Longhese, M. P., & Diffley, J. F. (2003). A central role for DNA replication forks in checkpoint activation and response. Molecular Cell, 11(5), 1323–1336. //doi.org/10.1016/S1097-2765(03)00169-2
Toledo, L. I., Murga, M., Zur, R., Soria, R., Rodriguez, A., Martinez, S., Oyarzabal, J., Pastor, J., Bischoff, J. R., & Fernandez-Capetillo, O. (2011). A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nature Structural & Molecular Biology, 18(6), 721. //doi.org/10.1038/nsmb.2076
Tsimaratou, K., Kletsas, D., Kastrinakis, N. G., Tsantoulis, P. K., Evangelou, K., Sideridou, M., Liontos, M., Poulias, I., Venere, M., Salmas, M., Kittas, C., Halazonetis, T. D., & Gorgoulis, V. G. (2007). Evaluation of claspin as a proliferation marker in human cancer and normal tissues. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland, 211(3), 331–339. //doi.org/10.1002/path.2095
Tuul, M., Kitao, H., Iimori, M., Matsuoka, K., Kiyonari, S., Saeki, H., Oki, E., Morita, M., & Maehara, Y. (2013). Rad9, Rad17, TopBP1 and claspin play essential roles in heat-induced activation of ATR kinase and heat tolerance. PLoS One, 8(2), e55361. //doi.org/10.1371/journal.pone.0055361
Walton, M. I., Eve, P. D., Hayes, A., Valenti, M. R., Alexis, K., Box, G., Hallsworth, A., Smith, E. L., Boxall, K. J., Lainchbury, M., Matthews, T. P., Jamin, Y., Robinson, S. P., Aherne, G. W., Reader, J. C., Chesler, L., Raynaud, F. I., Eccles, S. A., Collins, I., & Garrett, M. D. (2012). CCT244747 is a novel potent and selective CHK1 inhibitor with oral efficacy alone and in combination with genotoxic anticancer drugs. Clinical Cancer Research, 18(20), 5650–5661. //doi.org/10.1158/1078-0432.CCR-12-1322
Wang, C., Yang, C., Ji, J., Jiang, J., Shi, M., Cai, Q., Yu, Y., Zhu, Z., & Zhang, J. (2017). Deubiquitinating enzyme USP20 is a positive regulator of Claspin and suppresses the malignant characteristics of gastric cancer cells. International Journal of Oncology, 50(4), 1136–1146. //doi.org/10.3892/ijo.2017.3904
Wang, L., Wang, Y., Chen, A., Jalali, A., Liu, S., Guo, Y., Na, A., Nakshatri, H., Li, B. Y., & Yokota, H. (2018). Effects of a checkpoint kinase inhibitor, AZD7762, on tumor suppression and bone remodeling. International Journal of Oncology, 53(3), 1001–1012. //doi.org/10.3892/ijo.2018.4481
Wengner, A. M., Siemeister, G., Lücking, U., Lefranc, J., Wortmann, L., Lienau, P., Bader, B., Bömer, U., Moosmayer, D., Eberspächer, U., Golfier, S., Schatz, C. A., Baumgart, S. J., Haendler, B., Lejeune, P., Schlicker, A., von Nussbaum, F., Brands, M., Ziegelbauer, K., & Mumberg, D. (2020). The novel ATR inhibitor BAY 1895344 is efficacious as monotherapy and combined with DNA damage–inducing or repair–compromising therapies in preclinical cancer models. Molecular Cancer Therapeutics, 19(1), 26–38. //doi.org/10.1158/1535-7163.MCT-19-0019
Wu, M., Pang, J. S., Sun, Q., Huang, Y., Hou, J. Y., Chen, G., Zeng, J. J., & Feng, Z. B. (2019). The clinical significance of CHEK1 in breast cancer: A high-throughput data analysis and immunohistochemical study. International Journal of Clinical and Experimental Pathology, 12(1), 1.
Xiao, Y., Ramiscal, J., Kowanetz, K., Del Nagro, C., Malek, S., Evangelista, M., Blackwood, E., Jackson, P. K., & O’Brien, T. (2013). Identification of preferred chemotherapeutics for combining with a CHK1 inhibitor. Molecular Cancer Therapeutics, 12(11), 2285–2295. //doi.org/10.1158/1535-7163.MCT-13-0404
Xu, Y., Liu, Z., Sun, J., Pan, Q., Sun, F., Yan, Z., & Hu, X. (2011). Schisandrin B prevents doxorubicin-induced chronic cardiotoxicity and enhances its anticancer activity in vivo. PLoS One, 6(12), e28335. //doi.org/10.1371/journal.pone.0028335
Yang, C. C., Kato, H., Shindo, M., & Masai, H. (2019). Cdc7 activates replication checkpoint by phosphorylating the Chk1-binding domain of Claspin in human cells. eLife, 8, e50796. //doi.org/10.7554/eLife.50796
Yang, C. C., Suzuki, M., Yamakawa, S., Uno, S., Ishii, A., Yamazaki, S., Fukatsu, R., Fujisawa, R., Sakimura, K., Tsurimoto, T., & Masai, H. (2016). Claspin recruits Cdc7 kinase for initiation of DNA replication in human cells. Nature Communications, 7(1), 1–14. //doi.org/10.1038/ncomms12135
Yeeles, J. T., Janska, A., Early, A., & Diffley, J. F. (2017). How the eukaryotic replisome achieves rapid and efficient DNA replication. Molecular Cell, 65(1), 105–116. //doi.org/10.1016/j.molcel.2016.11.017
Yogosawa, S., & Yoshida, K. (2018). Tumor suppressive role for kinases phosphorylating p53 in DNA damage-induced apoptosis. Cancer Science, 109(11), 3376–3382. //doi.org/10.1111/cas.13792
Yoshizawa-Sugata, N., & Masai, H. (2007). Human Tim/Timeless-interacting protein, Tipin, is required for efficient progression of S phase and DNA replication checkpoint. Journal of Biological Chemistry, 282(4), 2729–2740. //doi.org/10.1074/jbc.M605596200
Yu, Q., La Rose, J., Zhang, H., Takemura, H., Kohn, K. W., & Pommier, Y. (2002). UCN-01 inhibits p53 up-regulation and abrogates γ-radiation-induced G2-M checkpoint independently of p53 by targeting both of the checkpoint kinases, Chk2 and Chk1. Cancer Research, 62(20), 5743–5748.
Yuan, J., Luo, K., Deng, M., Li, Y., Yin, P., Gao, B., Fang, Y., Wu, P., Liu, T., & Lou, Z. (2014). HERC2-USP20 axis regulates DNA damage checkpoint through Claspin. Nucleic Acids Research, 42(21), 13110–13121. //doi.org/10.1093/nar/gku1034
Zenke, F. T., Zimmermann, A., Dahmen, H., Elenbaas, B., Pollard, J., Reaper, P., Bagrodia, S., Spilker, M. E., Amendt, C., & Blaukat, A. (2019). Abstract 369: antitumor activity of M4344, a potent and selective ATR inhibitor, in monotherapy and combination therapy. Cancer Research. //doi.org/10.1158/1538-7445.am2019-369
Zhang, C., Yan, Z., Painter, C. L., Zhang, Q., Chen, E., Arango, M. E., Kuszpit, K., Zasadny, K., Hallin, M., Hallin, J., Wong, A., Buckman, D., Sun, G., Qiu, M., Anderes, K., & Christensen, J. G. (2009). PF-00477736 mediates checkpoint kinase 1 signaling pathway and potentiates docetaxel-induced efficacy in xenografts. Clinical Cancer Research, 15(14), 4630–4640. //doi.org/10.1158/1078-0432.CCR-08-3272
Zhao, H., Wang, Z., Zhu, M., Liao, J., & Xu, X. (2021). USP11 suppresses CHK1 activation by deubiquitinating CLASPIN. Genome Instability & Disease. //doi.org/10.1007/s42764-021-00034-1
Zhou, Z. R., Yang, Z. Z., Wang, S. J., Zhang, L., Luo, J. R., Feng, Y., Yu, X. L., Chen, X. X., & Guo, X. M. (2017). The Chk1 inhibitor MK-8776 increases the radiosensitivity of human triple-negative breast cancer by inhibiting autophagy. Acta Pharmacologica Sinica, 38(4), 513–523. //doi.org/10.1038/aps.2016.136
Zou, L. (2017). DNA replication checkpoint: New ATR activator identified. Current Biology, 27(1), R33–R35. //doi.org/10.1016/j.cub.2016.11.025
Acknowledgements
The works described from our laboratory were supported by JSPS KAKENHI Grant Number 20K21410 (to H.M.) and 19K16367 (to C-C. Y.) and by the funding from Hirose Foundation (to C-C. Y.).
Author information
Affiliations
Genome Dynamics Project, Department of Basic Medical Sciences, Tokyo Metropolitan Institute of Medical Science, Setagaya-ku, Tokyo, Japan
Hao-Wen Hsiao, Chi-Chun Yang & Hisao Masai
Department of Computational Biology and Medical Sciences, The University of Tokyo, Kashiwa-shi, Chiba, Japan
Hao-Wen Hsiao & Hisao Masai
Corresponding author
Correspondence to Hisao Masai.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit //creativecommons.org/licenses/by/4.0/.
About this article
Cite this article
Hsiao, HW., Yang, CC. & Masai, H. Roles of Claspin in regulation of DNA replication, replication stress responses and oncogenesis in human cells. GENOME INSTAB. DIS. (2021). //doi.org/10.1007/s42764-021-00049-8
Received10 July 2021
Revised19 August 2021
Accepted24 August 2021
Published07 September 2021
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
ATR-Claspin-Chk1
DNA replication
Replication stress
Replication checkpoint control
Oncogenesis
Biological stresses
用户登录
还没有账号?
立即注册